CASTEP
For this specific format, the minimum required files are:
A user defined input file (.param).
A cell file (.cell), with the atomic cooordinates, cell dimensions and kpoints used in the calculation.
The generated resume output file (.castep).
Note
CASTEP allows to change the default units used inside the calculation and printed in its output file. The current ioChem-BD release supports the default units only.
The geometry optimization method used must be BFGS, the default one. Damped molecular dynamics optimizations are not supported.
Geometry optimization calculations generate a .geom file that must be also appended when uploading it to ioChem-BD, otherwise the optimized geometry won’t be available in the final resume.
All generated CASTEP graph files .xcd can be attached to the upload process and its content will be rendered in the final resume.
General Info
Field |
Source |
Sample value |
|---|---|---|
Title |
Set on Browse calculation publication |
Sample calculation |
Browse Item |
URL pointing Browse published item |
|
Program |
CASTEP 2021 HF1 |
|
Author |
Username fullname |
Doe, John |
Calculation type |
Custom logic 1 |
Geometry optimization |

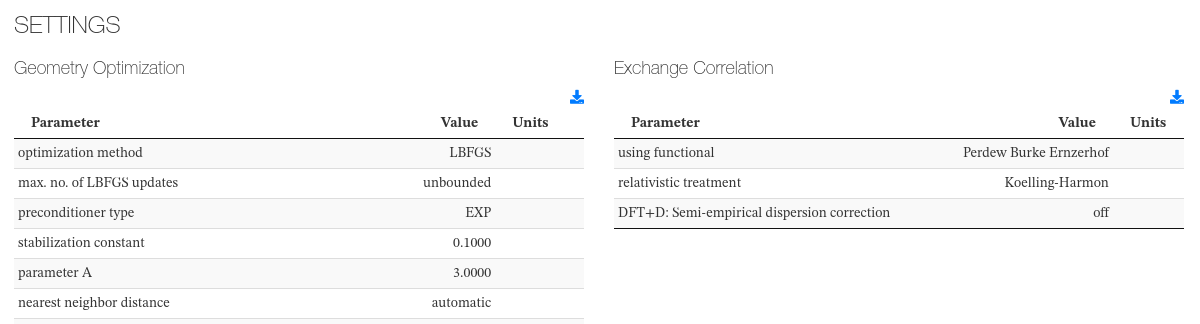
Settings
This section captures initialization settings like units used, geometry optimization parameters, exchange correlation parameters, etc.
Data source: <module cmlx:templateRef=’parameters’>
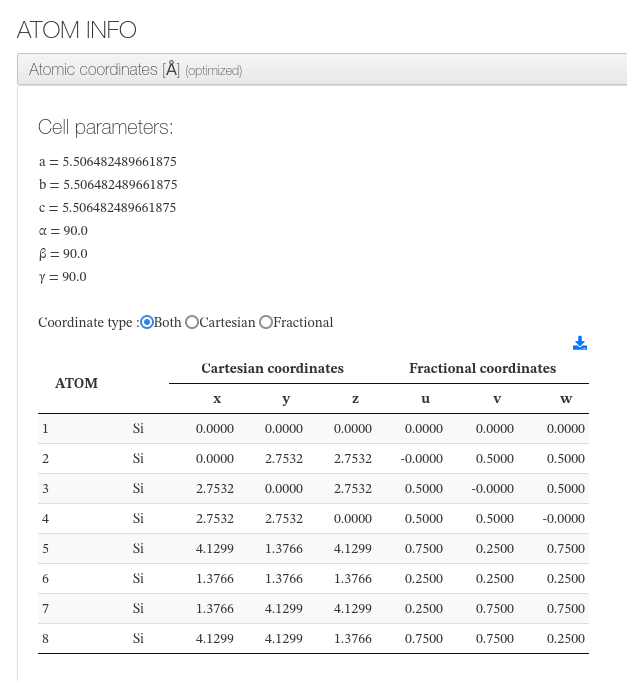
Atomic coordinates
After settings, our HTML resume will output cell dimensions and an xyz coordinates table with current molecule atoms.
For every atom, we will output it’s serial number, atom type, coordinates in angstroms in cartesian and fractional types.
Information is read from geometry steps file or from atoms section.
On geometry optimization calculations it will indicate if it converged along with the initial and final geometries.
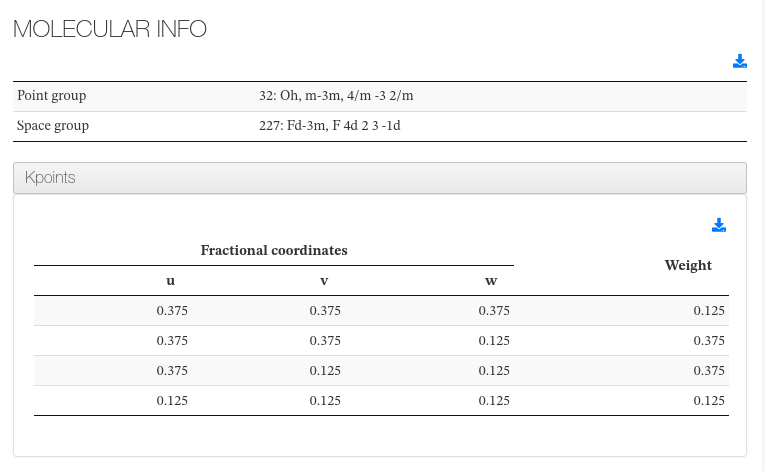
Molecular Info
This section captures molecule additional information not captured on previous section.
Field |
Source |
Description |
|---|---|---|
Point group |
<scalar dictRef=”cc:pointgroup”> |
Variable found inside Symmetry and Constraints section |
Space group |
<scalar dictRef=”ca:spacegroup”> |
Variable found inside Symmetry and Constraints section |
k-point |
Read from castep.cell |
K-point list and its weight, defined inside .cell file |
Modules
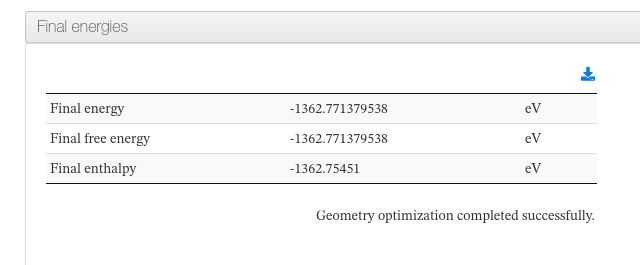
Energies
Data source:
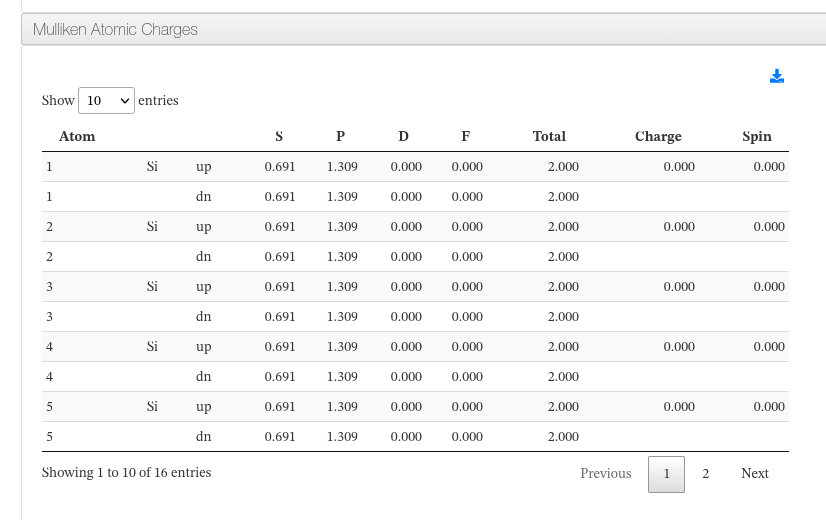
Mulliken Atomic Charges
Data source:
<module cmlx:templateRef=’mulliken’>
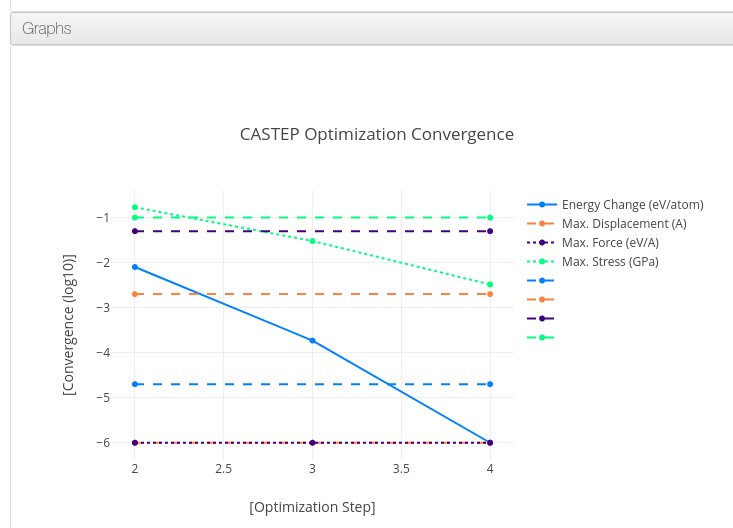
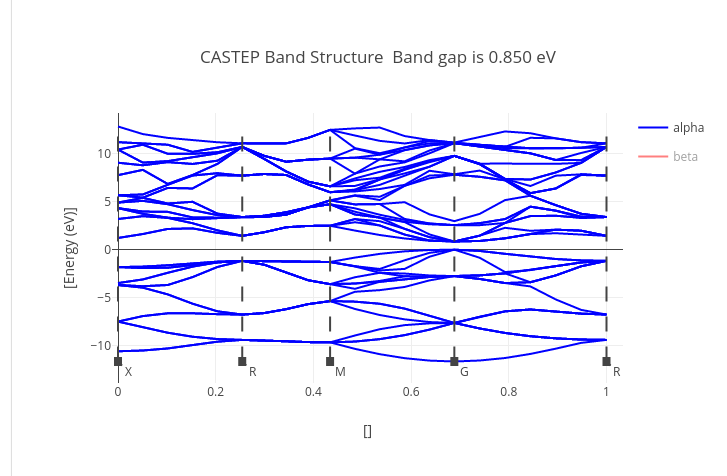
Graphs
Data source:
.xcd files attached to the calculation during its upload.
It can contain geometry optimization step parameters, density of states or band gap graphs.

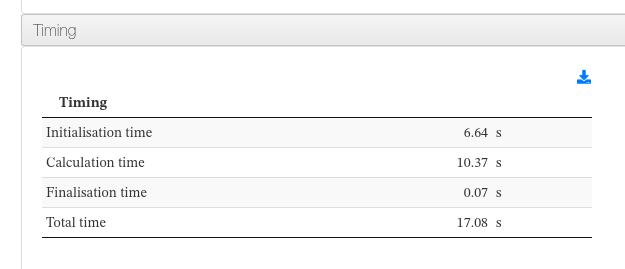
Timing
Data sources:
<module cmlx:templateRef=’timing’>
- 1
string
ca:getCalcTypestringsetup1 $setup Refers to all read elements from <module cmlx:templateRef="parameters"> 2 3 <xsl:variable name="ca:minimize">Geometry optimization</xsl:variable> 4 <xsl:variable name="ca:spectroscopy">Electronic spectroscopy</xsl:variable> 5 6 <xsl:function name="ca:getCalcType"> 7 <xsl:param name="setup" /> 8 9 <xsl:variable name="type" select="ca:getParameter($setup, 'type of calculation')" /> 10 <xsl:choose> 11 <xsl:when test="matches($type, 'geometry optimization')"> 12 <xsl:value-of select="$ca:minimize"/> 13 </xsl:when> 14 <xsl:when test="matches($type, 'Electronic Spectroscopy')"> 15 <xsl:value-of select="$ca:spectroscopy"/> 16 </xsl:when> 17 <xsl:otherwise><xsl:text>N/A</xsl:text></xsl:otherwise> 18 </xsl:choose> 19 </xsl:function> 20 21 <xsl:function name="ca:getParameter"> 22 <xsl:param name="setup" /> 23 <xsl:param name="name" /> 24 <xsl:copy-of select="$setup/cml:parameter/cml:scalar[@dictRef='x:label'][text()=$name]/following-sibling::cml:scalar[@dictRef='x:value']"/> 25 </xsl:function>