Shell automated scripts
All ioChem-BD shell client commands can be shell-scripted to ease upload files into the Create Module in an automated way. Find herein few helper scripts that will assist you when uploading calculations.
By using scripts, uploading data automatically is just a trivial exercise.
All our shell commands are contained within the shell client, which is downloadable from your Create web page. Feel free to customize these scripts to fit your needs, or turn them into new ones. Set the -v parameter (verbose) on these scripts to display more information about each script mechanism.
Per file type
loadadf
Loads an ADF calculation into the Create module (until AMS2020).
| Parameters | Description |
|---|---|
| -i filename | Input file, if not defined will use script defined name |
| -o filename | Output file, if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i and -o are not set, this script will look for input.in and output.out files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadadf script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.in"
OUTPUT_DEFAULT_FILENAME="output.out"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload calculation a-pw12 using a-pw12.opt.in and a-pw12.opt.out files
$ loadadf -i a-pw12.opt.in -o a-pw12.opt.out -n "a-pw12" -d "Optimization a-pw12"Upload calculation named metane using me-mp2.in and me-mp2.out files; name and description are picked from the parent folder name.
$ pwd
/home/user/Desktop/methane
$ loadadf -i me-mp2.in -o me-mo2.outUpload calculation named a-pw12.opt and attach additional file report.pdf
$ loadadf -i a-pw12.opt.in -o a-pw12.opt.out -a report.pdf -n a-pw12.opt -d "Optimization a-pw12"Upload calculation and automatically build its parent folder. calculation name and description will be same as parent folder
$ loadadf -i irc_ts2_09.in -o irc_ts2_09.out --autoloadams
Loads an AMS ADF calculation into the Create module (since AMS2020).
Data is typically retrieved from proprietary binary files (ams.rkf, adf.rkf) via two upload methods:
- by uploading the files themselves, or
- by using the rkf2cml tool to extract data into a single CML file
| Parameters | Description |
|---|---|
| -i filename | Input file, if not defined will use script defined name |
| -rkf1 filename | ams.rkf binary file, if not defined will use script defined name |
| -rkf2 filename | adf.rkf binary file, if not defined will use script defined name |
| -cml filename | CML file derived from .rkf binary files |
| -o filename | Output file, if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If the -cml parameter is used, the use of either -rkf1 or -rkf2 is forbidden, and viceversa.
If parameters -i, -rkf1, -rkf2 and -o are not set, this script will look for input.in, ams.rkf, adf.rkf and output.out files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadams script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.in"
OUTPUT_DEFAULT_FILENAME="output.out"
AMS_DEFAULT_FILENAME="ams.rkf"
ADF_DEFAULT_FILENAME="adf.rkf"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload calculation a-pw12 with its files:
$ loadams -i a-pw12.opt.in -o a-pw12.opt.out -rkf1 Test.results/ams.rkf -rkf2 Test.results/adf.rkf -n "a-pw12" -d "Optimization a-pw12"Upload calculation named a-pw12.opt and attach additional file report.pdf
$ loadams -i a-pw12.opt.in -o a-pw12.opt.out -rkf1 Test.results/ams.rkf -rkf2 Test.results/adf.rkf -a report.pdf -n a-pw12.opt -d "Optimization a-pw12"Upload calculation and automatically build its parent folder. calculation name and description will be same as parent folder
$ loadams -i irc_ts2_09.in -o irc_ts2_09.out -rkf1 Test.results/ams.rkf -rkf2 Test.results/adf.rkf --autoExamples
Instead of using .rkf files, its derived .cml can be used a posteriori (normally due to large .rkf files were removed).
User will generate the .cml file using the rkf2cml tool and later append it using -cml parameter.
$ rkf2cml ams.rkf adf.rkf rkf-derived.cml
...
$ # When calculation has to be uploaded, the .cml file must be provided
$ loadams -i irc_ts2_09.in -o irc_ts2_09.out -cml rkf-derived.cml -n a-pw12.opt -d a-pw12.optloadamber
Loads a Amber calculation into the Create module.
| Parameters | Description |
|---|---|
| -i filename | Input file, if not defined will use script defined name |
| -o filename | Output file, if not defined will use script defined name |
| -p filename | Parameter/topology file specification (.prmtop), if not defined will use script defined name |
| -ir filename | Initial coordinates file (.inpcrd) or intial restart file (.ncrst) used, if not defined will use script defined name |
| -r filename | Final restart file (.ncrst), if not defined will use script defined name |
| -t filename | Trajectory file (.nc), if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i, -o, -p, -ir, -r and/or -t are not set, this script will look for input.in, output.out, topology.prmtop, coords.inpcrd, restartnc.ncrst and trajectory.nc files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadamber script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.in"
OUTPUT_DEFAULT_FILENAME="output.out"
TOPOLOGY_DEFAULT_FILENAME="topology.prmtop"
COORDINATES_DEFAULT_FILENAME="coords.inpcrd"
RESTART_DEFAULT_FILENAME="restartnc.ncrst"
TRAJECTORY_DEFAUL_FILENAME="trajectory.nc"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
This command will set upload calculation name and description equals to parent folder and upload all files that match by name.
$ loadamberThis command will upload all matching files in directory, set calculation name as "Au-CoO-H2O" and description as "Sample upload".
$ loadamber -n Au-CoO-H2O -d "Sample upload"This command will behave the same as previous one but will upload different files rather than default.
$ loadamber -n Au-CoO-H2O -d "Sample upload" -i step1.in -o step1.out -p Au-CoO-H2O.prmtop -ir Au-CoO-H2O.inpcrd -r Au-CoO-H2O.ncrst -t Au-CoO-H2O.ncloadcastep
Loads a CASTEP calculation into the Create module.
More about the format capture restrictions on the following page.
| Parameters | Description |
|---|---|
| -i filename | Input parameters file (.param), if not defined will use script defined name |
| -o filename | Output resume file (.castep), if not defined will use script defined name |
| -oc filename | Cell file (.cell), if not defined will use script defined name |
| -xcd filename | Graph file (.xcd), can be repeated to add multiple files (optional) |
| -og filename | Geometry steps file (.geom), it is mandatory only on geometry optimization calculations. |
| -a filename | Append additional file, can be repeated to add multiple files (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section |
| |
If parameters -i, -o and -oc are not set, this script will look for calc.param, calc.castep, calc.cell files. If that files are missing, the upload process will be aborted.
If you wish to use another naming convention set the parameters to override default values or edit the loadcastep script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="calc.param"
OUTPUT_DEFAULT_FILENAME="calc.castep"
CELL_DEFAULT_FILENAME="calc.cell"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Additional files, like trajectories (.trj), can be attached to the uploaded calculation using the -a additional file parameter.
Examples
$ loadcastepThis command will set upload calculation name and description equals to parent folder and upload all required files if they match by name.
$ loadcastep -n Si_51688 -d "Sample upload"This command will upload required matching files in directory, set calculation name as "Si_51688" and description as "Sample upload".
$ loadcastep -n Si_51688 -d "Sample upload" -i Si_51688.param -o Si_51688.castep -oc Si_51688.cell -xcd Si_51688_Energies.xcd -xcd Si_51688_Convergence.xcdThis command will use different filenames and also upload two additional graph files.
$ loadcastep -n Si_51688 -d "Sample upload" -i Si_51688.param -o Si_51688.castep -oc Si_51688.cell -geom Si_51688.geomThis command will add the .geom file due to it's an optimization calculation.
$ loadcastep -n Si_51688 -d "Sample upload" -i Si_51688.param -o Si_51688.castep -oc Si_51688.cell -a Si_51688.trjThis command will upload the calculation adding a trajectory file as an additional file.
loadgauss
Same parameters and functionalities than the loadadf script.
WARNING
It is advised to use #p flag in Gaussian calculations. Link information helps ioChem-BD to capture some more extra information such as basis sets used.
loadgromacs
Loads a GROMACS calculation into the Create module.
| Parameters | Description |
|---|---|
| -i filename | Molecular dynamics parameters (.mdp) as the Input file, if not defined will use script defined name |
| -o filename | Logfile (.log) as the Output file, if not defined will use script defined name |
| -oc filename | Molecular structure in Gromos87 format. (.gro), if not defined will use script defined name |
| -t filename | Trajectory file (.xtc), if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i, -o, -oc and/or -t are not set, this script will look for input.mdp, output.log, geometry.gro and trajectory.xtc files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadgromacs script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.mdp"
OUTPUT_DEFAULT_FILENAME="output.log"
GEOMETRY_DEFAULT_FILENAME="geometry.gro"
TRAJECTORY_DEFAULT_FILENAME="trajectory.xtc"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload calculation LIN24_LI192 using required files
$ loadgromacs -i npt.mdp -o LIN24_LI192.log -oc LIN24_LI192.gro -t LIN24_LI192.xtc -n "LIN24_LI192" -d "LIN24 LI192 SOL25208 40000ps NPT 300K calculation"Upload same calculation but name and description are picked from the parent folder name.
$ pwd
/home/user/Desktop/LIN24_LI192
$ loadgromacs -i npt.mdp -o LIN24_LI192.log -oc LIN24_LI192.gro -t LIN24_LI192.xtcUpload calculation and automatically build its parent folder. calculation name and description will be same as parent folder
$ loadgromacs -i npt.mdp -o LIN24_LI192.log -oc LIN24_LI192.gro -t LIN24_LI192.xtc --autoloadgronor
Loads an GronOR calculation into the Create module.
| Parameters | Description |
|---|---|
| -i filename | Input file (optional) |
| -o filename | Output file in CML format, if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameter -o are not set, this script will look for output.out files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadgronor script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
OUTPUT_DEFAULT_FILENAME="output.cml"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
This command will set upload calculation name and description equals to parent folder and upload all files that match by name.
$ loadgronorThis command will upload all matching files in directory, set calculation name as "Sc2C82" and description as "Sample upload".
$ loadgronor -n Sc2C82 -d "Sample upload"This command will behave the same as previous one but will upload different files rather than default.
$ loadgronor -n Sc2C82 -d "Sample upload" -i input2.in -o dimmer.cmlloadgrrm
Same parameters and functionalities than the loadadf script.
WARNING
Reaction path searches are not yet supported (AFIR, etc).
loadlammps
Loads a LAMMPS calculation into the Create module.
Read more about LAMMPS format capture restricitions on the following page.
| Parameters | Description |
|---|---|
| -i filename | User defined input file, if not provided will use script defined name |
| -p filename | Data file used on the read_data command, if not defined will use script defined name |
| -o filename | Output file, called by default log.lammps, if not defined will use script defined name |
| -t filename | Trajectory file, it can be compressed in .zip or .tar.gz, if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i, -o, -p and/or -t are not set, this script will look for input.in, log.lammps, lammps.dat and trajectory.zip files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadlammps script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.in"
OUTPUT_DEFAULT_FILENAME="log.lammps"
DATAFILE_DEFAULT_FILENAME="lammps.dat"
TRAJECTORY_DEFAULT_FILENAME="trajectory.zip"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload calculation LIN24_LI192 using required files
$ loadlammps -i lammps.lmp -p lammps.dat -o log.lammps -t trajectory.zip -n "LIN24_LI192" -d "LIN24 LI192 SOL25208 40000ps NPT 300K calculation"Upload same calculation but name and description are picked from the parent folder name.
$ pwd
/home/user/Desktop/LIN24_LI192
$ loadlammps -i lammps.lmp -p lammps.dat -o log.lammps -t trajectory.zipUpload calculation and automatically build its parent folder. calculation name and description will be same as parent folder
$ loadlammps -i lammps.lmp -p lammps.dat -o log.lammps -t trajectory.zip --autoloadmolcas
Same parameters and functionalities than the loadadf script.
loadmopac
Same parameters and functionalities than the loadadf script.
loadorca
Loads an ORCA calculation into the Create module, it can be used standalone as other shell client commands or it can be attached to a job script to be automatically uploaded after the calculation has finished.
| Parameters | Description |
|---|---|
| -i filename | Input file, (optional), if not defined will use script defined name |
| -o filename | Output file (optional), if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| -molden | Build molden file with calculation molecular orbitals from the binary .gbw file. Will use orca_m2kl script to do such conversion. Please read -molden section for further configuration. |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i and -o are not set, it will look for input.in and 'output.out' files, if they are missing, the upload will be aborted. If we use another naming convention just edit the loadorca script file and replace default file names.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.in"
OUTPUT_DEFAULT_FILENAME="output.out"If parameters -n and -d are not defined, the loadorca script will use the parent folder's name as calculation name and description. If a parameter value contains multiple words and blank spaces (like description), they must be enclosed inside double quotes.
Molden orbitals file generation
Along with input, output and additional files. This script allows generating a molden-format file that contains calculation molecular orbitals. If we use this flag, uploaded calculations will also display molecular orbitals on JSmol viewer. To generate such file, we must first edit the loadorca script file that resides on the shell client folder. First lines define multiple orca_2mkl_X_X_X variables.
orca_2mkl_2_8="/opt/ORCA2_8/orca_x86_64_exe_r2131/orca_2mkl"
orca_2mkl_2_9_0="/opt/ORCA2_9/orca_x86_64_exe_r2131/orca_2mkl"
orca_2mkl_3_0_0=
orca_2mkl_3_0_1=We must set the correct path to this script file to match your file system configuration. orca_2mkl is installed along with Orca software. Be careful to define as much variables and paths for each installed Orca version on your system. If you use -molden parameter on a Orca v2.8 output file and its parameter is undefined, wrong or points a different version (2.9, 3.0, ...), it will fail to work.
Examples
Upload calculation ete using ete.inp and ete.out files
$ loadorca -i ete.inp -o ete.out -n "ete optimization" -d "Optimization ete"Upload calculation named ncs2 using ncs2.inp and ncs2.out files, name and description came from parent folder, then build molden molecular orbital files and upload it.
$ pwd
/home/user/Desktop/ncs2
$ loadorca -i ncs2.inp -o ncs2.out -moldenUpload calculation and automatically build its parent folder, calculation name and description will be same as parent folder
$ loadorca -i ete.inp -o ete.out --autoloadqespresso
Loads a QuantumESPRESSO calculation into the Create module.
Options like -b, -dos, -pi or -.po are restricted to specificic calculation types described below.
| Parameters | Description |
|---|---|
| -i filename | Input file, if not defined will use script defined name. |
| -o filename | Output file, if not defined will use script defined name. |
| -as filename | Absorption spectra data file, see Absorption spectra section below for further details (optional). |
| -b filename | Reordered band data file, see Band structure section below for further details (optional). |
| -dos filename | PDOS atom file(s) (optional). |
| -pi filename | Input used on the phonon matdyn.x tool, contains k-points and labels (optional). |
| -po filename | Output file containing the phonon frequencies. (optional) |
| -a filename | Append additional file (optional). |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name. |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name. |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i and -o are not set, this script will look for input.in and output.out files. If any of that files don't exist, the upload process will be aborted. If you wish to use another naming convention just edit the loadqespresso script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.in"
OUTPUT_DEFAULT_FILENAME="output.out"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload calculation a-pw12 using a-pw12.opt.in and a-pw12.opt.in files
$ loadqespresso -i a-pw12.opt.in -o a-pw12.opt.out -n "a-pw12" -d "Optimization a-pw12"Upload calculation named bencene using pw_scf.in and pw_scf.out files; name and description are picked from the parent folder name.
$ pwd
/home/user/Desktop/bencene
$ loadqespresso -i pw_scf.in -o pw_scf.outUpload calculation and automatically build its parent folder. calculation name and description will be same as parent folder
$ loadqespresso -i irc_ts2_09.in -o irc_ts2_09.out --autoAbsorption spectra calculations
ioChem-BD can process and display absorption spectrum data coming from time-dependent density functional theory calculations (TDDFT).
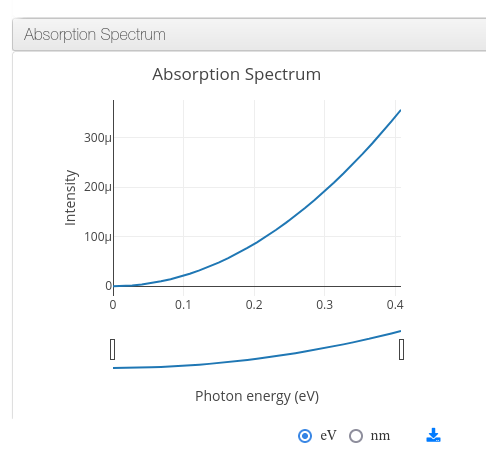
The required file is generated by the combination of the following QuantumEspresso tools:
pw.x -> turbo_lanczos.x -> turbo_spectrum.x
SCF -> TDDF calculation -> Spectrum creation
Once the spectrum data file is generated, it can be uploaded along with the calculation using the -as parameter.
Example
Upload calculation named a-pw12.opt and attach spectra file plot_chi.dat
$ loadqespresso -i pw_scf.in -o pw_scf.out -as plot_chi.dat -n a-pw12.opt -d "Bencene calculation"Band structure calculations
ioChem-BD can process and display band dispersion data on its HTML report.
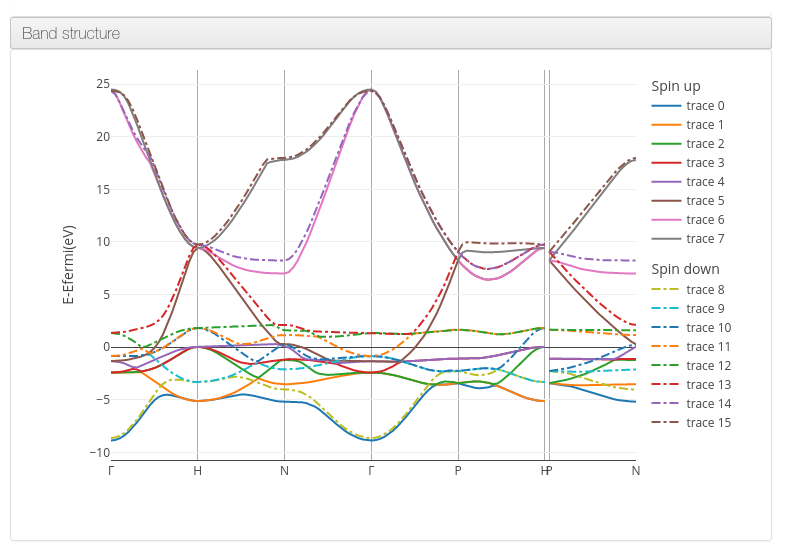
The following QuantumEspresso tool workflow will generate the required files:
pw.x -> pw.x (bands) -> bands.x
SCF -> Band path calculation -> Band dispersion calculation
Once the bands dispersion file is generated, it must be uploaded along with the calculation using the -b parameter. When having spin up/down band files, please provide two -b parameters, being the spin up band file the first and the down file the second.
The provided input file must be the band path calculation file from 2nd step, which contains kpoint band path.
The provided output files must be the SCF output file from the 1st step and the band path calculation file from 2nd step, so two -o parameters should be provided, the order must be kept the same as described here.
WARNING
The first output file (SCF) is optional but if not provided the generated graph won't center its energies on fermi energy value.
Examples
Upload calculation named Ce_bands and attach band structure information file bands.out:
$ loadqespresso -i ce_nscf.in -o ce_scf.out -o ce_nscf.out -b bands.out -n Ce_bands -d "Bands for Ce atom"Same calculation but with spin up/down band files:
$ loadqespresso -i ce_nscf.in -o ce_scf.out -o ce_nscf.out -b bands_up.out -b bands_down.out -n Ce_bands -d "Bands for Ce atom"Projected Density of States calculations
ioChem-BD can process and display the projected density of states information on its HTML report.
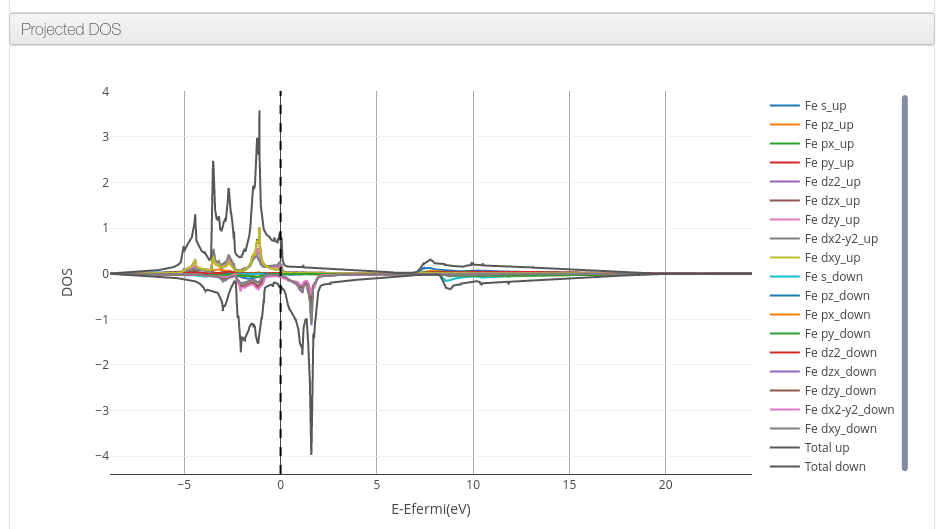
The following QuantumEspresso tool workflow will generate the required files:
SCF -> NSCF -> Projects wavefunctions onto orthogonalized atomic wavefunctions.
We need first a self-consistent field calculation and then a non-self-consistent field calculation with denser k-points. Then we prepare the input file for the tool projwfc.x.
The tool generates all the PDOS values files following this naming template:
.*.pdos_atm#XXX(YYY)_wfc#AAA(BBB)Being the first two parameters (in uppercase) the id and element symbol of the atom and the last two the id and number of the atomic orbital.
All the atom pdos files must be uploaded along with the calculation using the -dos parameter on each file, the totals dos file is not required because it is calculated from the individual values.
The provided output files must be the SCF output file from the 1st step and the NSCF output file from the 2nd step, so two -o parameters should be provided, the order must be kept the same as described here.
WARNING
Review the section DATA CONVERSION -> From CML to HTML -> QuantumEspresso to check the used file format.
Example
Upload calculation named CeO2_pdos and attach the pdos files:
$ loadqespresso -i ceo2_nscf.in -o ceo2_scf.out -o ceo2_nscf.out -n CeO2_pdos -d 'Cerium dioxide PDOS' -dos 'pwscf.pdos_atm#1(Fe)_wfc#1(s) '-dos pwscf.pdos_atm#1\(Fe\)_wfc#2\(p\) -dos 'pwscf.pdos_atm#1(Fe)_wfc#3(d)'On calculation with spin up/down files we will upload all PDOS files as on the previous example.
WARNING
The PDOS filename contain symbols that can break the upload process if they are not escaped or enclosed between single quotes, please review last the command for both examples.
Phonon dispersion
ioChem-BD can process and display phonon dispersion data on its HTML report.
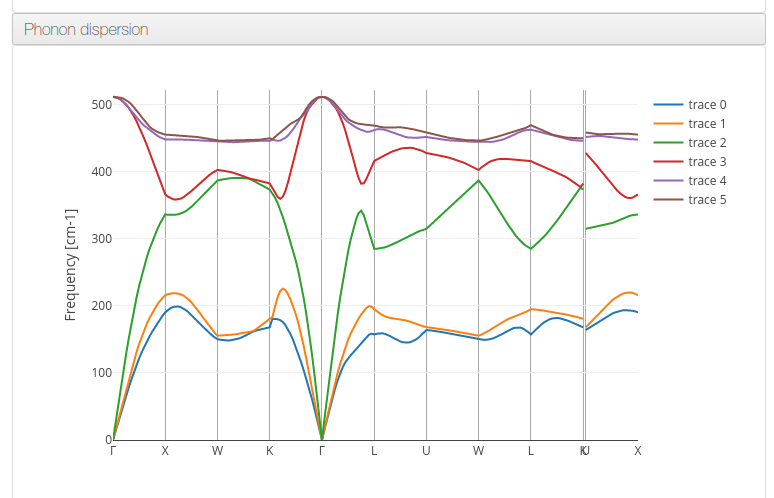
The following QuantumEspresso tool workflow will generate the required files:
Structure optimization -> PHonon -> Phonon dispersion calculation
Once the structure is relaxed, the dynamical matrix is calculated on the on 2nd step to finally get the phonon dispersion on the 3rd calculation.
The provided input -i and output file -o must be the optimization ones from 1st step.
The provided phonon input file -pi will contain the kpoint definition, weights and labels. Finally the -po will be the data file containing the phonon frequency level on each kpoint.
WARNING
Review the section DATA CONVERSION -> From CML to HTML-> QuantumEspresso to check the used file format.
Example
Upload calculation named Ce_phonon and attach phonon dispersion information file matdyn.freq:
$ loadqespresso -i ce_opt.in -o ce_opt.out -pi qe_matdyn_phdisp.pwi -po matdyn.freq -n Ce_phonon -d "Phonon dispersion for Ce atom"Phonon DOS
ioChem-BD can process and display phonon density of states data on its HTML report.
Will use the same tool workflow that the previous Phonon dispersion example but on the 3rd step will request matdyn.x tool to generate Phonon DOS data.
The uploaded files will be the same except that, in this case, the -pi file won't be required, just the the -po with the data file containing the phonon dos energies.
Example
Upload calculation named Ce_phonon_dos and attach phonon dispersion information file matdyn.dos:
$ loadqespresso -i ce_opt.in -o ce_opt.out -po matdyn.dos -n Ce_phonon_dos -d "Phonon DOS for Ce atom"loadturbo
Loads a Turbomole calculation into the Create Module. It can be used standalone as other shell client commands, or it can be attached to a job script to be automatically uploaded after the calculation has finished.
| Parameters | Description |
|---|---|
| -i control | Input file (optional) , if not defined it will look for control file on current folder |
| -o job.last | Output file (optional) , if not defined it will look for job.last file on current folder |
| -oe energy | Energy file (optional), if not defined it will look for energy file on current folder |
| -ob basis | Basis file (optional) , if not defined it will look for basis file on current folder |
| -oc coord | Coord file (optional), if not defined it will look for coord file on current folder |
| -a file | Append an additional file (optional) |
| -n | Name of the calculation in the data base (optional), if not defined it will use the parent folder's name |
| -d desc | Description of the calculation in the data base (optional), if not defined it will use parent folder's name |
| --auto | Autogenerate current path into the Create module (optional). Refer to -auto parameter section. |
If parameters -i and -o are not set, it will look by default for control and job.last files on current folders, if they are missing the upload will be aborted. If we use another naming convention just edit loadturbo script file and replace the default file names.
# Default static file names, change them in order to fit your naming conventions
CONTROL_DEFAULT_FILENAME="control"
ENERGY_DEFAULT_FILENAME="energy"
BASIS_DEFAULT_FILENAME="basis"
COORD_DEFAULT_FILENAME="coord"
JOB_LAST_DEFAULT_FILENAME="job.last"If parameters -n and -d are not defined, the loadturbo script will use the parent folder's name as calculation name and description. If a parameter value contains multiple words and blank spaces (like description), they have to be enclosed inside double quotes.
Multiple step calculation
On calculations that generate multiple output files like dscf, escf, we can upload them as a unique file by joining its output files into one. We do so by repeating the -o parameter with the name of each output file. Must be defined in the same order as they were generated. Example:
$ loadturbo -i control -o dscf.out -o escf.outThe output of freeh (the free enthalpy module), can also be appended also using the -o parameter. HTML Report will then display thermochemistry values and energy corrections.
$ loadturbo -i control -o job.last -o freeh.outWARNING
The freeh module processing is only valid for single temperature and pressure values, it will not work for ranges.
Examples
Upload calculation flourophenol using control. job.last, energy, basis and coord files. Set name as flourophenol.
$ loadturbo -i control -o job.last -n "flourophenol" -d "Population analysis" -oe energy -ob basis -oc coordWe can omit multiple parameters if they are named the same as default script names, so the last command can be the same than this one:
$ loadturbo -n "flourophenol" -d "Population analysis"If no parameter is set, name and description fields will come from parent folder.
$ pwd
/home/user/Desktop/flourophenol
$ loadturboloadsiesta
Loads a SIESTA calculation into the Create module.
For this chemical format, the provided output file must not be the textual one, it must be the XML one. It is generated by declaring the following flag on the input file:
XML.Write .true.| Parameters | Description |
|---|---|
| -i filename(s) | Input file(s), if not defined will use script defined name |
| -o filename | Output file, if not defined will use script defined name |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
SIESTA can combine multiple input files to bundle the final input file, in this case, you can set multiple -i parameters, one for each partial input file.
If parameters -i and -o are not set, this script will look for input.fdf and output.xml files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadsiesta script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="input.fdf"
OUTPUT_DEFAULT_FILENAME="output.xml"If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload h2o calculation, that contains two input files and a output .xml file:
$ loadsiesta -n h2o -d \"Sample upload\" -i h2o.fdf -i Default.fdf -o h2o.xmlloaduma
Loads a UMA-ASE (Universal Model for Atoms + Atomistic Simulation Environment) calculation into the Create module.
Read more about the format and its software at its GitLab project <https://gitlab.com/carlesbo/uma-ase>.
The calculation data is provided as a single .zip bundle file. Format detection is performed automatically by inspecting the bundle contents, so no separate input file is required.
| Parameters | Description |
|---|---|
| -o filename | UMA-ASE zip bundle file (required) |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
The -o parameter is mandatory; unlike other load scripts, there is no default filename fallback. If it is not provided the upload process will be aborted.
If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
Upload calculation uma-opt using uma-opt.zip bundle file
$ loaduma -o uma-opt.zip -n "uma-opt" -d "UMA-ASE geometry optimisation"Upload calculation with name and description picked from the parent folder name.
$ pwd
/home/user/Desktop/uma-opt
$ loaduma -o uma-opt.zipUpload calculation and attach additional file report.pdf
$ loaduma -o uma-opt.zip -a report.pdf -n uma-opt -d "UMA-ASE geometry optimisation"Upload calculation and automatically build its parent folder. Calculation name and description will be same as parent folder.
$ loaduma -o uma-opt.zip --autoloadvasp
Loads a VASP calculation into the Create module, it can be used standalone as other shell client commands or it can be attached to a job script to be automatically uploaded after the calculation has finished.
| Parameters | Description |
|---|---|
| -i INCAR | Input file, if not defined it will look for INCAR file on current folder |
| -o OUTCAR | Output file, set to OUTCAR by default |
| -c CONTCAR | CONTCAR file containing final geometry coordinates (required for single file uploads, not needed for NEB/DIM) |
| -dc DOSCAR | Attach DOSCAR file information to resulting uploaded calculation (optional), it will extract Density of states coefficients. |
| -kp KPOINTS | Attach KPOINTS file information to resulting uploaded calculation (optional), it will extract KPOINT generation data |
| -a filename | Coord file (optional), if not defined it will look for the coord file from the current folder |
| -n | Name of the calculation in the data base (optional), if not defined the calculations' output file name will be used |
| -d desc | Description of the calculation in the data base (optional), if not defined the calculations' file name will used. |
| --auto | Autogenerate current path into Create module (optional). Refer to the -auto parameter section. |
| -neb | Upload Nudged Elastic Band calculation, see more at: Uploading NEB/DIM calculations section |
| -dim | Upload Dimmer calculation, see more at: Uploading NEB/DIM calculations section |
Examples
Uploads a calculation and sets identical name and description as the parent folder. It will capture INCAR, OUTCAR and CONTCAR files by default
$ loadvaspAutomatically generates parent folder structure and uploads calculation and sets identical name and description as the parent folder. It will capture INCAR, OUTCAR and CONTCAR files by default, and will attach report.pdf file to calculation.
$ loadvasp -a report.pdf --autoUploads calculation and associates its KPOINTS and DOSCAR files, sets name and description to opt1. It will capture INCAR, OUTCAR and CONTCAR files by default
$ loadvasp -n opt1 -d opt1 -kp KPOINTS -dc DOSCARUpload calculation set name and description to opt2. Will capture INCAR2 and OUTCAR2 files, because we set -i and -o parameters.
$ loadvasp -i INCAR2 -o OUTCAR2 -n opt2 -d opt2 -kp KPOINTS -dc DOSCARUploading VASP NEB/DIM calculation
NEB
The nudged elastic band (NEB) is a method for finding saddle points and minimum energy paths between known reactants and products. The method works by optimizing a number of intermediate images along the reaction path.1 This kind of calculation generates multiple output files (images) inside a collection of numbered subfolders. To upload this kind of calculations we will set -neb parameter on base folder. loadvasp script will expect at last this file structure, it will iterate all XX folders and concatenate all outcar files.
.
├── 00
│ └── OUTCAR
├── 01
│ └── OUTCAR
├── 02
│ └── OUTCAR
├── 03
│ └── OUTCAR
├── 04
│ └── OUTCAR
├── 05
│ └── OUTCAR
├── 06
│ └── OUTCAR
└── INCARExample of a NEB calculation upload: Upload NEB, attach KPOINTS file information and set NEB-1 as name and description.
$ loadvasp -neb -kp KPOINTS -n NEB-1-2 -d NEB-1-2DIM
The dimer method is one of the min-mode following methods that allows the user to start from any initial configuration and search for a nearby saddle point. This method can also be used to start from a minimum basin and search in random directions for saddle points.2 We can just upload calculation as a normal single calculation or we can use -dim parameter if we already have initial and final state calculations, so we must set a folder structure like this:
.
├── IS
│ └── OUTCAR
├── TS
│ ├── INCAR
│ └── OUTCAR
└── FS
└── OUTCARExample of a DIM calculation upload: Upload Dimmer, attach KPOINTS and DOSCAR information and set NO_dim as name and description.
$ loadvasp -dim -kp KPOINTS -dc TS/DOSCAR -n NO_dim -d NO_dimloadxtb
Loads an xTB (extended tight-binding) calculation into the Create module.
| Parameters | Description |
|---|---|
| -i filename | Input control file (required), if not defined will use script defined name |
| -g filename | Geometry/coordinate file (required), if not defined will use script defined name |
| -o filename | Output log file (required), if not defined will use script defined name |
| -f filename | Gaussian frequency output file (g98.out) (optional). Auto-detected if present in current directory. |
| -a filename | Append additional file (optional) |
| -n name | Name of the calculation in the data base (optional), if not defined will use parent folder name |
| -d desc | Description of the calculation in the data base (optional), if not defined will use parent folder name |
| --auto | Autogenerate current path into Create module (optional). Refer to -auto parameter section. |
If parameters -i, -g and -o are not set, this script will look for xtbinput.inp, coord.xyz and output.log files. If that info is missing, the upload process will be aborted. If you wish to use another naming convention just edit the loadxtb script file and replace default filenames.
# Default static file names, change them in order to fit your naming conventions
INPUT_DEFAULT_FILENAME="xtbinput.inp"
GEOMETRY_DEFAULT_FILENAME="coord.xyz"
OUTPUT_DEFAULT_FILENAME="output.log"
FREQUENCY_DEFAULT_FILENAME="g98.out"The -f parameter is optional; if not specified and g98.out exists in the current directory, it will be uploaded automatically. This file is generated by xTB when running frequency calculations with the --ohess or --hess flags.
If parameters -n and -d are not defined, the scripts will use the parent folder's name as calculation name and description.
Examples
This command will set upload calculation name and description equals to parent folder and upload all files that match by name.
$ loadxtbThis command will upload all matching files in directory, set calculation name as "water" and description as "Water geometry optimization".
$ loadxtb -n water -d "Water geometry optimization"This command will behave the same as previous one but will upload different files rather than default.
$ loadxtb -n water -d "Water optimization" -i water.inp -g water.xyz -o water.logUpload frequency calculation including the Gaussian frequency output file.
$ loadxtb -n water -d "Water frequencies" -i water.inp -g water.xyz -o water.log -f g98.outAdditional commands
--auto parameter
In some situations our calculations lay inside a complex folder structure, under a high number of nested subfolders. Such project structure can be generated via the Create web interface or using the Create shell client commands, like cpro, cdpro and pwdpro. This can be a time-consuming process, so we can use the -auto parameter to generate such structure for us. Imagine that we want to upload a calculation inside a path from a folder mounted like this:
/home/user/mnt_cluster/metOH-oxidation/MoO2/metOH/TS1_NEB/02/reopt/freqGenerating such project hierarchy via shell commands (excluding $HOME) will be like this
$ cpro -n metOH-oxidation -d metOH-oxidation
$ cdpro metOH-oxidation
$ cpro -n MoO2 -d MoO2
$ cdpro MoO2
$ cpro -n metOH -d metOH
$ cdpro metOH
$ cpro -n TS1_NEB -d TS1_NEB
$ cdpro TS1_NEB
$ cpro -n 02 -d 02
$ cdpro 02
$ cpro -n reopt -d reopt
$ cdpro reopt
$ cpro -n freq -d freq
$ cdpro freq
$ loadgauss -i input.in -o output -n freq1 -d freq1Using -auto parameter can be as simple as this
$ cd /home/user/mnt_cluster/metOH-oxidation/MoO2/metOH/TS1_NEB/02/reopt/freq
$ loadgauss -i input.in -o output -n freq1 -d freq1 **-auto**Generated projects path will generate also mnt_cluster project, because -auto parameter excludes $HOME. If we want to exclude mnt_cluster folder or our folder is mounted in another path that does not contain a $HOME path like /mnt/cluster/... we can edit our upload script loadXXXX:
#On auto mode we will generate a project path, navigate inside and then upload calculation
if [ $auto -gt 0 ]; then
moveBasePath
full_path="$(cd "$(dirname "$output")"; pwd)" #Will use 'output' file parent folders
home_path="$(cd $HOME_PATH/mnt_cluster; pwd)" #Set this home_path if uploading from /home/user/mnt_cluster
home_path="$(cd /mnt/cluster; pwd)" #Set this home_path if uploading from /mnt/cluster
partial_path="${full_path/$home_path/''}" #Remove user home folder
partial_path="${partial_path//[-@\$\%\& \"]/_}" #Replace special characters and blank spaces by underscore
if [ $verbose -gt 0 ]; then
echo "Generating path : "$partial_path
fi
projects=$(echo $partial_path | tr "/" "\n")
for project in $projects
do
if [ $project = "''" -o $project = "" ]; then
continue
fi
project="${project//[-@\$\%\&\"\' ]/_}"
createpro="cpro -n "$project" -d "$project
changepro="cdpro "$project
executeRepCommand "$REP_SCRIPTS/exe-rep-command $changepro" "" #Try cdpro to project, if it fails, we'll create this project
if [ ! $retval -eq 0 ]; then
executeRepCommand "$REP_SCRIPTS/exe-rep-command $createpro" ""
executeRepCommand "$REP_SCRIPTS/exe-rep-command $changepro" ""
fi
done
figetproject
This script is an example of how the shell client tools can be used in order to retrieve, query or manipulate content that is inside Create module.
It retrieves a complete project content using the shell client commands. Project names will be prepended with p_ and calculations with c_ to ease its grouping and reading.
Must be run using the source command to be effective. The project full path must be provided as parameter, user must be the owner of that project or at least have read rights on it.
$ source getproject /db/username/hexenolTo execute it outside the shell client folder, append the path to the shell client to your PATH environment variable.
Shell upload automation
We can attach these lines to our calculation job scripts, so after the calculation software finishes its processing, it gets uploaded automatically into ioChem-BD, making this step totally unattended.
Consider adding more verification steps inside upload scripts to avoid uploading erroneous calculations like premature exits, program crashes, not converged optimizations, etc.
In these examples, we consider that the shell client resides on $HOME/shell:
Gaussian job submit file example
#!/bin/bash
#\` -pe smp\* 12
#\$ -N test
#\$ -cwd
input=input.in
output=output.out
/home/programs/bin/gaussian09.sh C01 $input $output
. $HOME/shell/start-rep-shell
loadgauss -i $input -o $output -n $output -d $output --auto
exit-repNumbered lines do:
- Connect to Create
- Upload calculation using Gaussian script and $input and $output parameters (builds path automatically)
- Disconnect from Create
Turbomole job submit file example
#!/bin/bash
#$ -pe smp* 1-
#$ -cwd
#$ -q mag14.q
. /home/programs/bin/turbomole.sh 6.6
export TURBOTMPDIR=$TMPDIR
cp HILL/* .
/usr/bin/time -o time_output dscf
output=$(basename $SGE_STDOUT_PATH)
. $HOME/shell/start-rep-shell
loadturbo -o $output --auto
exit-repNumbered lines do:
- Connect to Create
- Upload calculation using turbomole script and $output parameters
- Disconnect from Create
Recursive upload
We can take advantage of shell scripting to automate calculation upload of multiple nested folders and subfolders.
One calculation per folder
This is an example script that uploads a group of ADF calculations starting from current folder.
Restrictions:
- There must be only one input and ouput file inside each folder,
- Uploaded calculations will have its name same as its parent folder name.
We can customize which input and output files are captured setting wildcards *.in and *.out.
#!/bin/bash
. start-rep-shell # <---Please append full path to start-rep-shell command
for folder in $(find `pwd` -type d); do
echo "Processing folder :" $folder
inputfile="$(find $folder -maxdepth 1 -name '*.in' -printf "%f\n")"
outputfile="$(find $folder -maxdepth 1 -name '*.out' -printf "%f\n")"
if [ -z $inputfile ]; then
echo " Not matching files"
else
echo " Input file "$inputfile
echo " Output file "$outputfile
cd $folder
loadadf --auto -i $inputfile -o $outputfile
fi
done
exit-repWe can derive previous script into more sophisticated versions.
Multiple calculations per folder
The following script navigates inside all child folders, it search for all input and output files inside each folder and then uploads them. This script allows setting file extensions for input and output files.
Restrictions:
- Input and output files of the same calculation must have same name (and different extension)
- Uploaded calculation will be named as input file (to avoid name collisions)
#!/bin/bash
#Replace this variables to suit your naming convention
INPUT_FILE_EXTENSION=in
OUTPUT_FILE_EXTENSION=out
. start-rep-shell # <--- Please append full path to start-rep-shell command
for folder in $(find `pwd` -type d); do
echo "Processing folder :" $folder
for inputfile in $(find $folder -maxdepth 1 -name "*.$INPUT_FILE_EXTENSION" -printf "%f\n"); do
filename=$(echo $inputfile | sed "s/\.$INPUT_FILE_EXTENSION//")
outputfile="$filename.$OUTPUT_FILE_EXTENSION"
if [ -z $outputfile ]; then
echo " Not matching output file "$outputfile
else
cd $folder
loadgauss --auto -i $inputfile -o $outputfile -n $filename -d $filename
fi
done
done
exit-rep