XTB
XTB
XTB (extended tight-binding) is a semiempirical quantum chemistry program based on density functional tight-binding (DFTB) methods. It provides fast geometry optimizations, molecular dynamics, and property predictions for molecular systems using GFN-xTB models.
General Info
XTB - General Info - Main fields
| Field | Description | Example |
|---|---|---|
| Title | Calculation title, assigned during publication | Sample calculation |
| Browse Item | Link to the published item in the Browse repository | https://iochem-bd.iciq.es/browse/handle/100/5672 |
| Program | XTB program version used for the calculation | xtb 6.5.1 (git b0914e9) |
| Author | Full name of the user who submitted the calculation | Doe, John |
| Formula | Molecular formula in Hill notation, derived from the final geometry | C 6 H 6 |
| Calculation type | Custom logic [^1] | Geometry optimization |
| Method | GFN-xTB model variant used for the calculation | GFN2-xTB |
| Solvation model | Solvent used in the calculation (only shown when solvation is enabled) | water |
| Optimization | Convergence status of the geometry optimization (only shown for optimization runs) | Converged |
XTB - General Info - HTML example

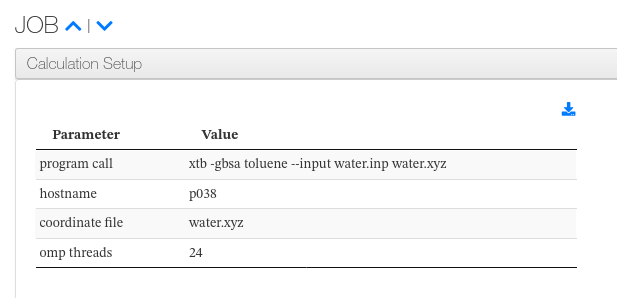
Calculation Setup
For each job, a collapsible panel lists the calculation setup parameters parsed from the XTB run header. Each row displays the parameter name alongside its configured value.
Calculation Setup - Displayed fields
| Column | Description |
|---|---|
| Parameter | Name of the setup parameter (e.g. program call, method, charge) |
| Value | Configured value for the parameter |
Calculation Setup - HTML example
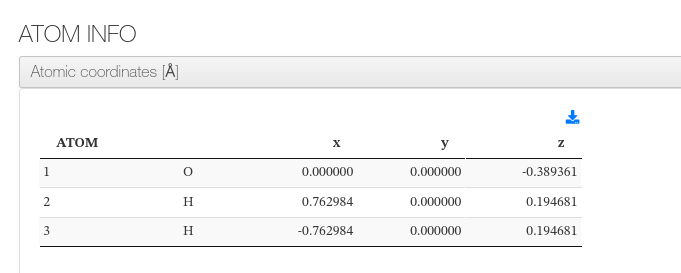
Atom Info
Displays a table of atomic coordinates in ångströms (Å). The final geometry is shown when available; otherwise the initial structure is displayed. A download button allows exporting the geometry in XYZ format.
Columns: atom index, element symbol, x, y, z (Å).
XTB - Atom Info - HTML example
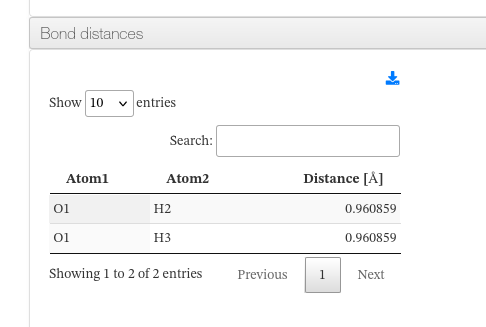
Bond Distances
When bond information is available, a collapsible panel displays the pairwise bond distances computed from the Cartesian coordinates. Each row identifies the two bonded atoms (element symbol + serial number) and the distance in ångströms (Å).
Bond distances - HTML example
Molecular Info
Shows molecular-level electronic properties of the system.
Molecular Info - Main fields
| Field | Description |
|---|---|
| Charge | Total net electrical charge of the system |
| Spin polarization | Number of unpaired electrons (only shown when present) |
| Multiplicity | Spin multiplicity, computed as spin polarization + 1 (only shown when spin polarization is present) |
Molecular Info - HTML example

Modules
When a calculation contains multiple jobs, each is shown as a collapsible section with the subsections below (when applicable).
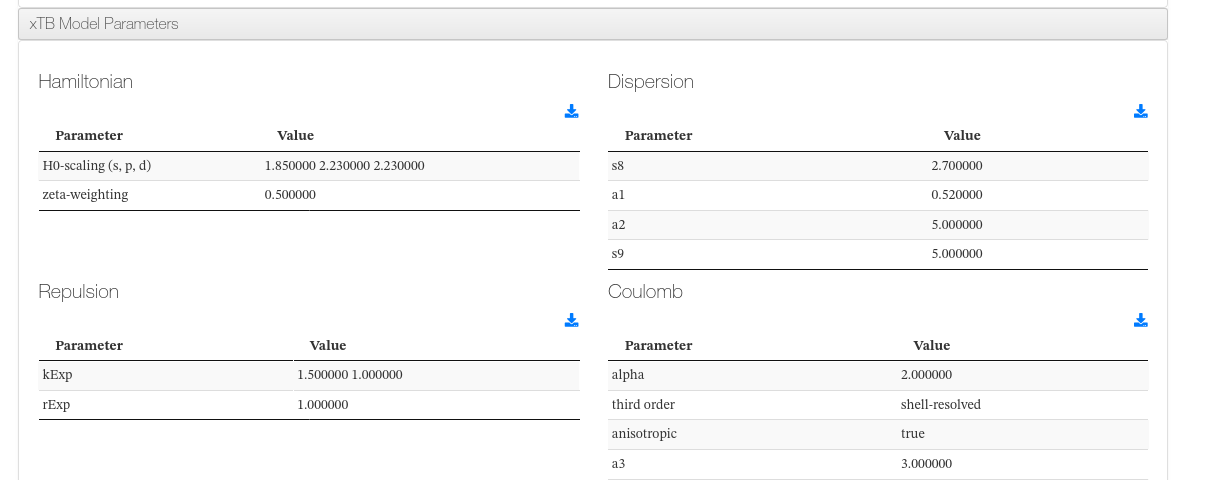
xTB Model Parameters
A collapsible panel displays the xTB model parameters grouped by section (Hamiltonian, Dispersion, Repulsion, Coulomb, Solvation, etc.). Each group is rendered as a separate sub-table with two columns: parameter name and value.
xTB Model Parameters - Common groups
| Group | Description |
|---|---|
| Hamiltonian | Method and Hamiltonian settings (e.g. GFN2-xTB) |
| Dispersion | Dispersion correction parameters |
| Repulsion | Repulsive potential parameters |
| Coulomb | Coulomb interaction parameters |
| Solvation | Implicit solvation model parameters (when solvation is enabled) |
xTB Model Parameters - HTML example
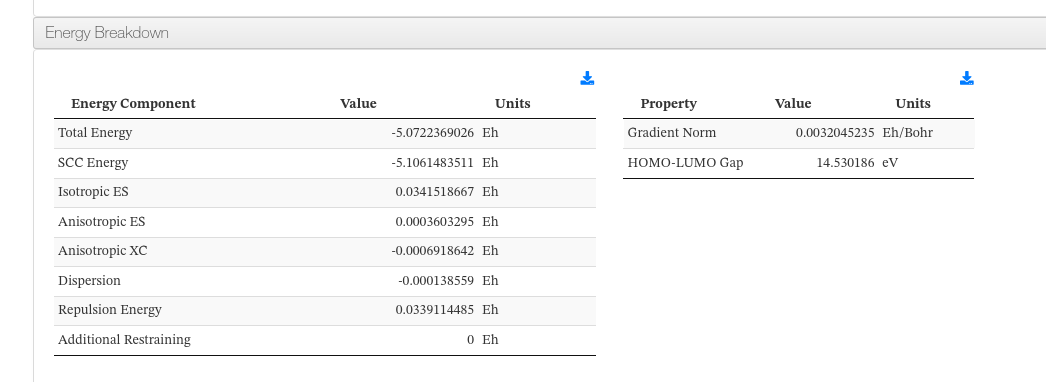
Energy Breakdown
Presents the total energy and its individual contributions from the final summary of the calculation. A second table shows the gradient norm and HOMO-LUMO gap. All energy values are displayed in Hartree (Eh).
Energy Breakdown - Main fields
| Field | Description |
|---|---|
| Total Energy | Total electronic energy of the system (Eh) |
| SCC Energy | Self-consistent charge contribution (Eh) |
| Isotropic ES | Isotropic electrostatic energy (Eh) |
| Anisotropic ES | Anisotropic electrostatic energy (Eh) |
| Anisotropic XC | Anisotropic exchange-correlation energy (Eh) |
| Dispersion | Dispersion correction energy (Eh) |
| Repulsion Energy | Repulsive pair-potential energy (Eh) |
| Additional Restraining | Restraint energy contribution (Eh) |
| Gradient Norm | Maximum gradient norm, indicating convergence quality (Eh/bohr) |
| HOMO-LUMO Gap | Energy gap between the highest occupied and lowest unoccupied molecular orbitals (eV) |
Energy Breakdown - HTML example
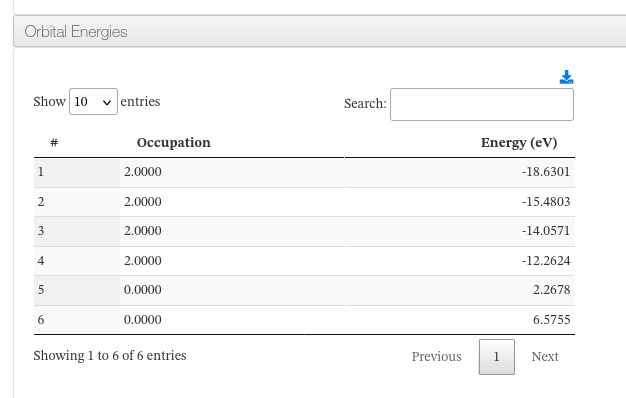
Orbital Energies
A collapsible panel lists the molecular orbital energies. For each orbital, the occupation number and energy are shown. The table is sortable and paginated.
Orbital Energies - Table columns
| Column | Description |
|---|---|
| # | Orbital index number |
| Occupation | Electron occupation number of the orbital |
| Energy | Orbital energy in electronvolts (eV) |
Orbital Energies - HTML example
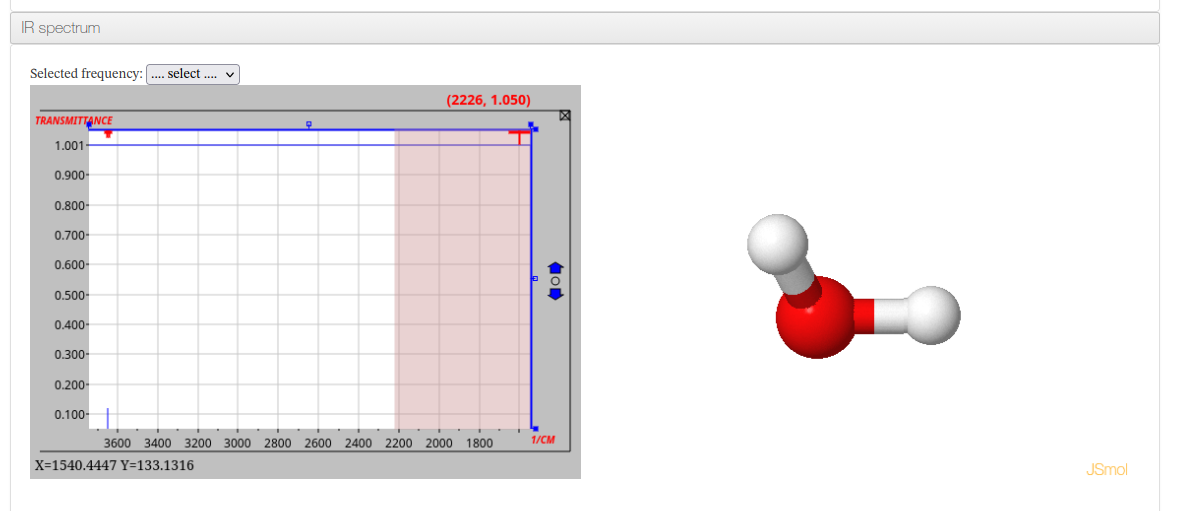
IR Spectrum
Shown when vibrational frequency data is available. Displays an interactive IR spectrum alongside a molecular viewer synchronized to the selected vibrational mode. A dropdown allows selecting individual frequencies.
IR Spectrum - HTML example
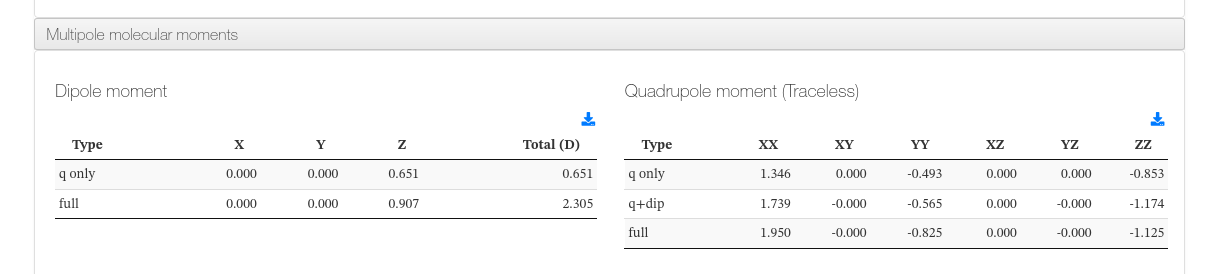
Multipole Moments
When available, a collapsible panel presents the dipole and quadrupole moments of the molecule.
Dipole Moment - Main fields
| Field | Description |
|---|---|
| q only (X, Y, Z) | Charge-only contribution to the dipole moment along each axis (Debye) |
| q only total (D) | Total charge-only dipole magnitude (Debye) |
| full (X, Y, Z) | Full dipole moment (charge + orbital contributions) along each axis (Debye) |
| full total (D) | Total dipole moment magnitude including all contributions (Debye) |
Quadrupole Moment (traceless) - Main fields
| Field | Description |
|---|---|
| q only | Charge-only contribution to the traceless quadrupole tensor (XX, XY, YY, XZ, YZ, ZZ) |
| q+dip | Charge + dipole contribution to the traceless quadrupole tensor |
| full | Full quadrupole tensor including all contributions |
Multipole Moments - HTML example
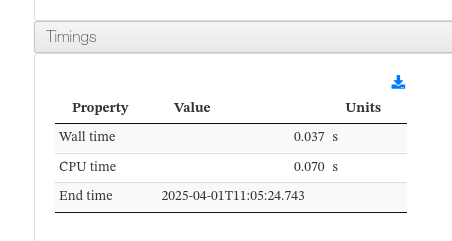
Timing
Reports the wall-clock time, CPU time, and completion timestamp of the calculation.
Timing - Main fields
| Field | Description |
|---|---|
| Wall time | Total elapsed wall-clock time for the calculation |
| CPU time | Total CPU time consumed across all cores |
| End time | Date and time when the calculation finished |
Timing - HTML example
[^1]: string xtb:getCalcType boolean hasOptimization boolean isConverged
<xsl:variable name="xtb:SinglePoint" select="'Single point'" />
<xsl:variable name="xtb:GeometryOptimization" select="'Geometry optimization'" />
<xsl:variable name="xtb:GeometryOptimizationNotConverged" select="'Geometry optimization (not converged)'" />
<xsl:variable name="xtb:FrequencyCalculation" select="'Frequency calculation'" />
<xsl:variable name="xtb:SinglePointHessian" select="'Single point Hessian (SPH)'" />
<xsl:variable name="xtb:MolecularDynamics" select="'Molecular dynamics'" />
<xsl:variable name="xtb:MetaDynamics" select="'Meta-dynamics'" />
<xsl:function name="xtb:getCalcType" as="xs:string*">
<xsl:param name="cml" as="node()"/>
<!-- Get program call from calculation.setup module -->
<xsl:variable name="programCall" as="xs:string?"
select="$cml//cml:module[@cmlx:templateRef='calculation.setup']
//cml:list[./cml:scalar[@dictRef='xtb:name']='program call']
/cml:scalar[@dictRef='xtb:value']"/>
<xsl:choose>
<!-- Check for metadynamics flag (\-\-metad) -->
<xsl:when test="contains($programCall, '--metad')">
<xsl:sequence select="$xtb:MetaDynamics"/>
</xsl:when>
<!-- \-\-omd: geometry optimization + molecular dynamics -->
<xsl:when test="contains($programCall, '--omd')">
<xsl:sequence select="xtb:getOptimizationStatus($cml)"/>
<xsl:sequence select="$xtb:MolecularDynamics"/>
</xsl:when>
<!-- \-\-md: molecular dynamics only -->
<xsl:when test="contains($programCall, '--md')">
<xsl:sequence select="$xtb:MolecularDynamics"/>
</xsl:when>
<!-- \-\-ohess: geometry optimization + frequency calculation -->
<xsl:when test="contains($programCall, '--ohess')">
<xsl:sequence select="xtb:getOptimizationStatus($cml)"/>
<xsl:sequence select="$xtb:FrequencyCalculation"/>
</xsl:when>
<!-- \-\-hess or \-\-freq: frequency calculation only -->
<xsl:when test="contains($programCall, '--hess') or contains($programCall, '--freq')">
<xsl:sequence select="$xtb:FrequencyCalculation"/>
</xsl:when>
<!-- \-\-opt: geometry optimization only, or fallback via ANCOPT section -->
<xsl:when test="contains($programCall, '--opt') or
exists($cml//cml:module[@cmlx:templateRef='optimization'])">
<xsl:sequence select="xtb:getOptimizationStatus($cml)"/>
</xsl:when>
<!-- Default: Single Point calculation -->
<xsl:otherwise>
<xsl:sequence select="$xtb:SinglePoint"/>
</xsl:otherwise>
</xsl:choose>
</xsl:function>
<!--
Helper function to get optimization status.
Returns the appropriate calculation type string based on convergence.
-->
<xsl:function name="xtb:getOptimizationStatus" as="xs:string">
<xsl:param name="cml" as="node()"/>
<xsl:variable name="convergedValue" as="xs:string?"
select="$cml//cml:scalar[@dictRef='xtb:converged']"/>
<xsl:choose>
<!-- Converged optimization -->
<xsl:when test="$convergedValue = 'converged'">
<xsl:sequence select="$xtb:GeometryOptimization"/>
</xsl:when>
<!-- Not converged optimization -->
<xsl:when test="$convergedValue = 'notconverged'">
<xsl:sequence select="$xtb:GeometryOptimizationNotConverged"/>
</xsl:when>
<!-- ANCOPT present but convergence scalar missing - assume converged -->
<xsl:when test="exists($cml//cml:module[@cmlx:templateRef='optimization'])">
<xsl:sequence select="$xtb:GeometryOptimization"/>
</xsl:when>
<!-- Fallback to single point -->
<xsl:otherwise>
<xsl:sequence select="$xtb:SinglePoint"/>
</xsl:otherwise>
</xsl:choose>
</xsl:function>